Parenteral Preparations, Challenges in Formulation
Published May 31, 2026
Presented at the 33rd Symposium on Particulate Preparations and Designs October 27-28, 2016, Nagano, Japan.
Author(s)
Blouet Elham
Pharm.D. - Head of Marketing|Biopharma - Injectables, Dialysis and Specialty APIs, Roquette
Introduction
Parenteral preparations are defined as solutions, suspensions, emulsions for injection or infusion, powders for injection or infusion, gels for injection and implants.1 They are sterile preparations intended to be administered directly into the systemic circulation in human or animal body.
They are required, like any pharmaceutical dosage forms, to meet the pharmaceutical quality standards as described in pharmacopeias and to be safe for the intended purpose of use.1, 2, 3 In addition to being sterile, parenteral preparations must be pyrogen-free. Sterility can be achieved by different processes of sterilization that should be appropriate to the formulations,4 while the pyrogen-free aspect will require, if no depyrogenation process is used during the preparation of the sterile drug products, the use of pyrogen-free pharmaceutical ingredients, drug substances or API (Active Pharmaceutical Ingredient) and excipients.
They are usually supplied in single dose glass or plastic containers (PVC nowadays less recommended, or polyolefin) or more and more in prefilled syringes or pens to facilitate the ease of use.1 This article will describe the main challenges encountered during the formulation of parenteral preparations, as well as Roquette’s solutions meeting the formulator’s needs.
Properties of parenteral preparations
The parenteral preparations are intended to be administered through the human or animal body, either by direct injections (for example, bolus intravenous [IV], intramuscular [IM] or subcutaneous [SC]) or by infusion with a controlled infusion rate or by direct implantation through IM or SC.
They must meet the following minimum compendia criteria.1, 2, 3
- To be sterile and pyrogen-free
- To be clear or practically exempt of visible particle and to be free from subvisible particles as required by pharmacopeias EP, USP and JP
- No evidence of phase separation for the emulsions, or aggregates formation for aqueous dispersion such as injectable Mab (monoclonal antibody) preparations
- In case of suspensions, the use of appropriate particle size and any sediment should be readily dispersed upon shaking to give stable formulations and ensure the correct dose to be withdrawn and injected.
Parenteral preparations may require the use of excipients that should be biocompatible, be selected for the appropriate use and to be included at the minimum efficient concentration.3 The functionality of these excipients is as follows:
- To make the preparations isotonic with respect to blood (glucose/dextrose, mannitol, sodium chloride…)
- To adjust the pH to the physiological one (mineral or organic acids or salts)
- To prevent the degradation of the drug substances (stabilizer…)
- To ensure or increase the drug substance’s solubility
- To provide adequate antimicrobial preservative property (only applicable to multidose preparations)
- It should be stressed that excipients should not adversely affect the intended medicinal action of the drug products, nor at the concentration used to cause toxicity or undue local irritation.
Challenges in formulations
The main challenge of all the different parenteral dosage forms is to achieve a good compatibility of the drug substances with the excipients (no formation of new impurities either by degradation of the drug substance or formation of new chemical entity between the drug substance and the excipients) as well as the compatibility of the preparations with the primary container (no leachable or adsorption to container).3
With regards to solutions and emulsions, the drug substances should be soluble and remain soluble during the entire shelf life of the drug products. When drug substances are not soluble, dissolution can be achieved by the use for instance either co-solvents, or surfactants, or a soluble pro-drug, or eventually the use of solubility enhancers such as the cyclodextrins thanks to the formation of inclusion complex.
The pH is one of the critical aspects of parenteral preparations which should have a pH close to the physiological one. However, in certain cases, a compromise should be found between the pH ensuring stability of the drug substance (such for peptides requiring alkaline pH or proteins at pH close to the isoelectric point) and the physiological one. In all cases, large volumes preparations (LVP, i.e., more than 100 ml as defined in pharmacopeia) should not contain a pH buffer as the blood has already a buffer effect property that could enter into competition with the injected drug product.
The stability of the drug substance is another critical point that a formulator can faced during the development of the formulation. Unstable drug substances will lead to the formation of new impurities jeopardizing the safety of use of the preparations. When the use of a stabilizer is justified (for instance the use of mannitol as free-radical scavenger or cysteine in paracetamol solution for injection), it should be included at the minimum concentration demonstrated to be efficient at release and during the entire shelf life3
In the cases of powders for injection or infusion obtained through a freeze-drying process, the use of bulking agent (such mannitol) and/or a cryoprotector will be needed when the dose of drug substance(s) cannot ensure solely the formation of acceptable “cake.”
Finally, the process of the sterilization should be selected according to the characteristics of the parenteral preparations (for instance, heat steam sterilization for aqueous solutions and dry heat for non-aqueous solutions), but in any cases, it can be justified by the nature of the primary containers.4 The efficiency of the selected sterilization process should be demonstrated through validation studies, using the appropriate biological indicators, to ensure an ASL (assurance sterility level) of 10-6.1
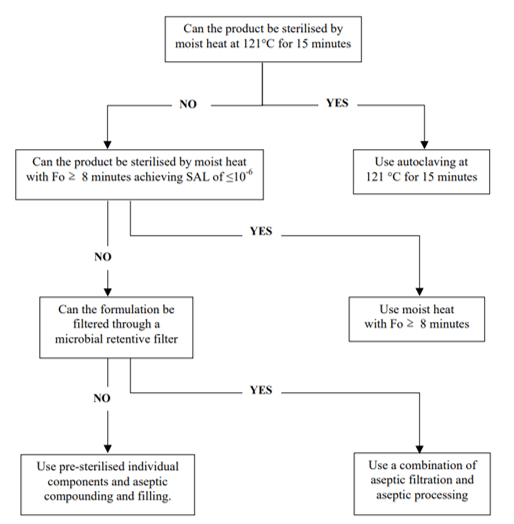
Figure 1. Decision tree for sterilization choices for aqueous products4
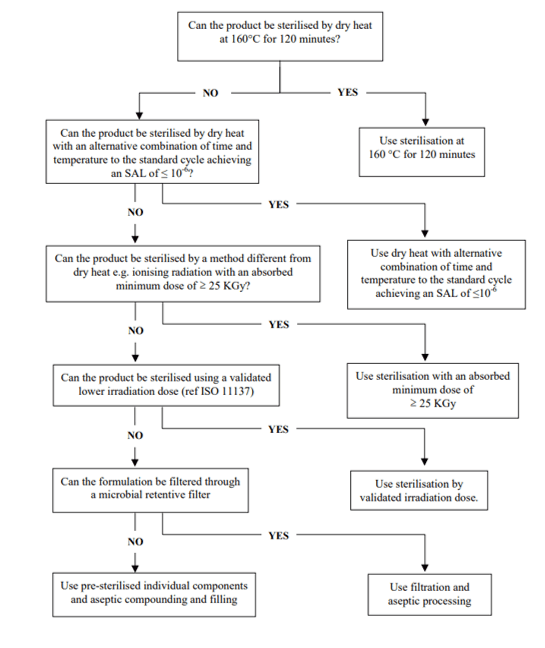
Figure 2. Decision tree for sterilization choices for non-aqueous liquid, semi-solid or dry powder products4
Roquette’s solutions5
Roquette has developed a pyrogen-free range of products with high pharmaceutical standards and being biocompatible for the manufacture of parenteral preparations. LYCADEX® PF (pyrogen-free dextrose monohydrate) is used as source of carbohydrates in large volume and small volume preparations (LVP and SVP) and in parenteral nutrition (TPN) as well as osmotic agent for dialysis solutions. PEARLITOL® PF (pyrogen-free mannitol) is used in LVP and SVP as APIs and isotonic agent, as well as bulking agent for freeze-dried injectable powders. NEOSORB® PF (pyrogen-free sorbitol) is also used as source of carbohydrates in LVP and SVP as well as osmotic agent in sterile irrigating fluids. KLEPTOSE® HPB and HP parenteral grade (pyrogen-free hydroxypropyl beta-cyclodextrin) is widely used as solubility and stability enhancer of APIs as well as enhancer of clinical tolerance. Sodium gluconate pharma is usually used as source of biological organic salts and as pH regulator.
All these pyrogen-free range of products are obtained from natural and renewable raw materials. Besides their compliance to pharmacopeias and other ICH quality requirements (for instance ICHQ3D for elemental impurities), all these pyrogen-free products, even when used as excipients, are manufactured in compliance to GMP, ICHQ7, and certified by competent authorities (ANSM the French Competent Authority and US FDA).
Conclusion
Parenteral preparations are sterile and pyrogen-free preparations intended to be administered directly into the systemic circulation in human or animal body. They should meet the pharmaceutical quality standards as described in pharmacopeias and ICH guidelines and also to ensure the clinical tolerance as well as to be safe for the intended purpose of use.
Roquette, thanks to its know-how and gained experience by serving for decades the big players of the injectable and dialysis markets, has developed a range of pyrogen-free products that meet all the required pharmaceutical standards and which are GMP/ICH Q7 certified whether they are used as APIs or excipients.
References
1) EP, USP and JP Pharmacopeias
2) European Medicines Agency, ICH Q6, Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products
3) European Medicines Agency, ICH Q8, Pharmaceutical development
https://www.ema.europa.eu/en/ich-q8-r2-pharmaceutical-development
4) European Medicines Agency, Decision trees for the selection of sterilization methods (CPMP/QWP/054/98)
https://www.ema.europa.eu/en/documents/scientific-guideline/superseded-annex-note-guidance-development-pharmaceutics-decision-trees-selection-sterilisation_en.pdf
5) Roquette Pharma Solutions
https://www.roquette.com/pharma
® Registered trademark(s) of Roquette Frères.
The information contained in this document is to the best of our knowledge true and accurate, but all instructions, recommendations or suggestions are made without any guarantee. Since the conditions of use are beyond our control, we disclaim any liability for loss and/or damage suffered from use of these data or suggestions. Furthermore, no liability is accepted if use of any product in accordance with these data or suggestions infringes any patent. No part of this document may be reproduced by any process without our prior written permission. For questions about a product’s compliance with additional countries’ standards not listed above, please contact your local Roquette representative.